

Construction prediction is a basic downside in molecular science as a result of the construction of a molecule determines its properties and capabilities. In recent times, deep studying strategies have made exceptional progress and influence on predicting molecular buildings, particularly for protein molecules. Deep studying strategies, reminiscent of AlphaFold and RoseTTAFold, have achieved unprecedented accuracy in predicting probably the most possible buildings for proteins from their amino acid sequences and have been hailed as a recreation changer in molecular science. Nonetheless, this methodology gives solely a single snapshot of a protein construction, and construction prediction can’t inform the entire story of how a molecule works.
Proteins are usually not inflexible objects; they’re dynamic molecules that may undertake totally different buildings with particular chances at equilibrium. Figuring out these buildings and their chances is important in understanding protein properties and capabilities, how they work together with different proteins, and the statistical mechanics and thermodynamics of molecular methods. Conventional strategies for acquiring these equilibrium distributions, reminiscent of molecular dynamics simulations or Monte Carlo sampling (which makes use of repeated random sampling from a distribution to attain numerical statistical outcomes), are sometimes computationally costly and will even grow to be intractable for advanced molecules. Due to this fact, there’s a urgent want for novel computational approaches that may precisely and effectively predict the equilibrium distributions of molecular buildings from primary descriptors.
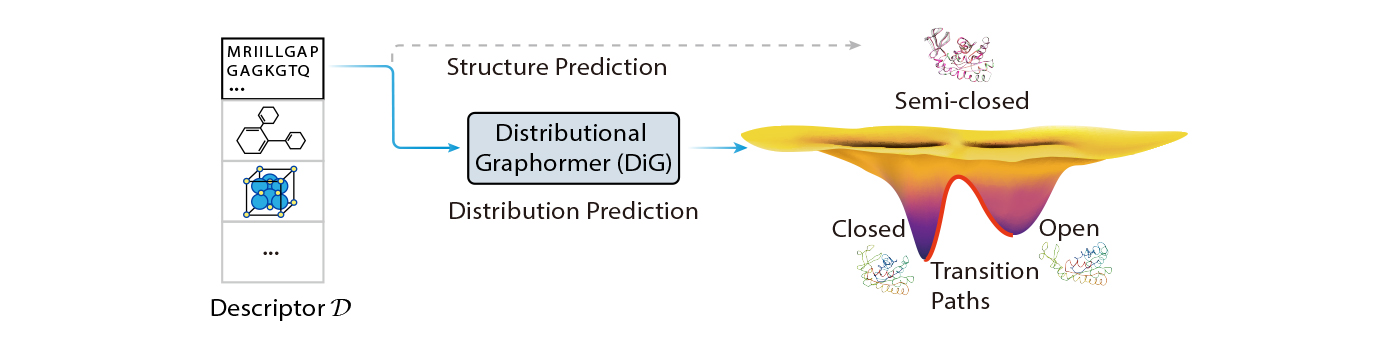
On this weblog publish, we introduce Distributional Graphormer (DiG), a brand new deep studying framework for predicting protein buildings in accordance with their equilibrium distribution. It goals to handle this basic problem and open new alternatives for molecular science. DiG is a major development from single construction prediction to construction ensemble modeling with equilibrium distributions. Its distribution prediction functionality bridges the hole between the microscopic buildings and the macroscopic properties of molecular methods, that are ruled by statistical mechanics and thermodynamics. However, this can be a great problem, because it requires modeling advanced distributions in high-dimensional house to seize the possibilities of various molecular states.
DiG achieves a novel resolution for distribution prediction by an development of our earlier work, Graphormer, which is a general-purpose graph transformer that may successfully mannequin molecular buildings. Graphormer has proven glorious efficiency in molecular science analysis, demonstrated by purposes in quantum chemistry and molecular dynamics simulations, as reported in our earlier weblog posts (see right here and right here for extra particulars). Now, now we have superior Graphormer to create DiG, which has a brand new and highly effective functionality: utilizing deep neural networks to straight predict goal distribution from primary descriptors of molecules.
Highlight: Microsoft Analysis Podcast
AI Frontiers: AI for well being and the way forward for analysis with Peter Lee
Peter Lee, head of Microsoft Analysis, and Ashley Llorens, AI scientist and engineer, focus on the way forward for AI analysis and the potential for GPT-4 as a medical copilot.
DiG tackles this difficult downside. It’s based mostly on the thought of simulated annealing, a basic methodology in thermodynamics and optimization, which has additionally motivated the current improvement of diffusion fashions that achieved exceptional breakthroughs in AI-generated content material (AIGC). Simulated annealing produces a posh distribution by steadily refining a easy distribution by the simulation of an annealing course of, permitting it to discover and settle in probably the most possible states. DiG mimics this course of in a deep studying framework for molecular methods. AIGC fashions are sometimes based mostly on the thought of diffusion fashions, that are impressed by statistical mechanics and thermodynamics.
DiG can also be based mostly on the thought of diffusion fashions, however we deliver this concept again to thermodynamics analysis, making a closed loop of inspiration and innovation. We think about scientists sometime will be capable to use DiG like an AIGC mannequin for drawing, inputting a easy description, reminiscent of an amino acid sequence, after which utilizing DiG to shortly generate lifelike and various protein buildings that observe equilibrium distribution. This can tremendously improve scientists’ productiveness and creativity, enabling novel discoveries and purposes in fields reminiscent of drug design, supplies science, and catalysis.
How does DiG work?
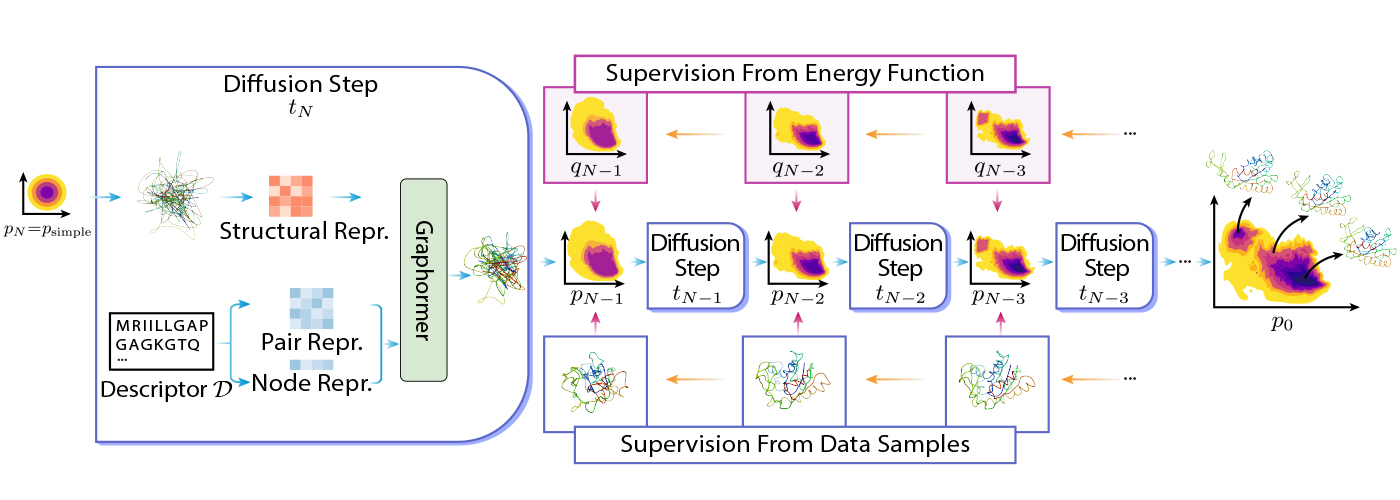
DiG relies on the thought of diffusion by reworking a easy distribution to a posh distribution utilizing Graphormer. The straightforward distribution could be a commonplace Gaussian, and the advanced distribution could be the equilibrium distribution of molecular buildings. The transformation is completed step-by-step, the place the entire course of mimics the simulated annealing course of.
DiG could be skilled utilizing various kinds of information or data. For instance, DiG can use vitality capabilities of molecular methods to information transformation, and it may possibly additionally use simulated construction information, reminiscent of molecular dynamics trajectories, to be taught the distribution. Extra concretely, DiG can use vitality capabilities of molecular methods to information transformation by minimizing the discrepancy between the energy-based chances and the possibilities predicted by DiG. This strategy can leverage the prior data of the system and practice DiG with out stringent dependency on information. Alternatively, DiG may also use simulation information, reminiscent of molecular dynamics trajectories, to be taught the distribution by maximizing the probability of the info below the DiG mannequin.
DiG reveals equally good generalizing skills on many molecular methods in contrast with deep learning-based construction prediction strategies. It’s because DiG inherits some great benefits of superior deep-learning architectures like Graphormer and applies them to the brand new and difficult job of distribution prediction. As soon as skilled, DiG can generate molecular buildings by reversing the transformation course of, ranging from a easy distribution and making use of neural networks in reverse order. DiG may also present the likelihood estimation for every generated construction by computing the change of likelihood alongside the transformation course of. DiG is a versatile and normal framework that may deal with various kinds of molecular methods and descriptors.
Outcomes
We show DiG’s efficiency and potential by a number of molecular sampling duties protecting a broad vary of molecular methods, reminiscent of proteins, protein-ligand complexes, and catalyst-adsorbate methods. Our outcomes present that DiG not solely generates lifelike and various molecular buildings with excessive effectivity and low computational prices, nevertheless it additionally gives estimations of state densities, that are essential for computing macroscopic properties utilizing statistical mechanics. Accordingly, DiG presents a major development in statistically understanding microscopic molecules and predicting their macroscopic properties, creating many thrilling analysis alternatives in molecular science.
One main utility of DiG is to pattern protein conformations, that are indispensable to understanding their properties and capabilities. Proteins are dynamic molecules that may undertake various buildings with totally different chances at equilibrium, and these buildings are sometimes associated to their organic capabilities and interactions with different molecules. Nonetheless, predicting the equilibrium distribution of protein conformations is a long-standing and difficult downside because of the advanced and high-dimensional vitality panorama that governs likelihood distribution within the conformation house. In distinction to costly and inefficient molecular dynamics simulations or Monte Carlo sampling strategies, DiG generates various and functionally related protein buildings from amino acid sequences at a excessive pace and a considerably decreased price.
DiG can generate a number of conformations from the identical protein sequence. The left aspect of Determine 3 reveals DiG-generated buildings of the principle protease of SARS-CoV-2 virus in contrast with MD simulations and AlphaFold prediction outcomes. The contours (proven as strains) within the 2D house reveal three clusters sampled by in depth MD simulations. DiG generates extremely related buildings in clusters II and III, whereas buildings in cluster I are undersampled. In the best panel, DiG-generated buildings are aligned to experimental buildings for 4 proteins, every with two distinguishable conformations akin to distinctive purposeful states. Within the higher left, the Adenylate kinase protein has open and closed states, each nicely sampled by DiG. Equally, for the drug transport protein LmrP, DiG additionally generates buildings for each states. Right here, notice that the closed state is experimentally decided (within the lower-right nook, with PDB ID 6t1z), whereas the opposite is the AlphaFold predicted mannequin that’s per experimental information. Within the case of human B-Raf kinase, the most important structural distinction is localized within the A-loop area and a close-by helix, that are nicely captured by DiG. The D-ribose binding protein has two separated domains, which could be packed into two distinct conformations. DiG completely generated the straight-up conformation, however it’s much less correct in predicting the twisted conformation. Nonetheless, in addition to the straight-up conformation, DiG generated some conformations that look like intermediate states.
One other utility of DiG is to pattern catalyst-adsorbate methods, that are central to heterogeneous catalysis. Figuring out energetic adsorption websites and steady adsorbate configurations is essential for understanding and designing catalysts, however it’s also fairly difficult because of the advanced surface-molecular interactions. Conventional strategies, reminiscent of density purposeful idea (DFT) calculations and molecular dynamics simulations, are time-consuming and dear, particularly for big and complicated surfaces. DiG predicts adsorption websites and configurations, in addition to their chances, from the substrate and adsorbate descriptors. DiG can deal with varied forms of adsorbates, reminiscent of single atoms or molecules being adsorbed onto various kinds of substrates, reminiscent of metals or alloys.
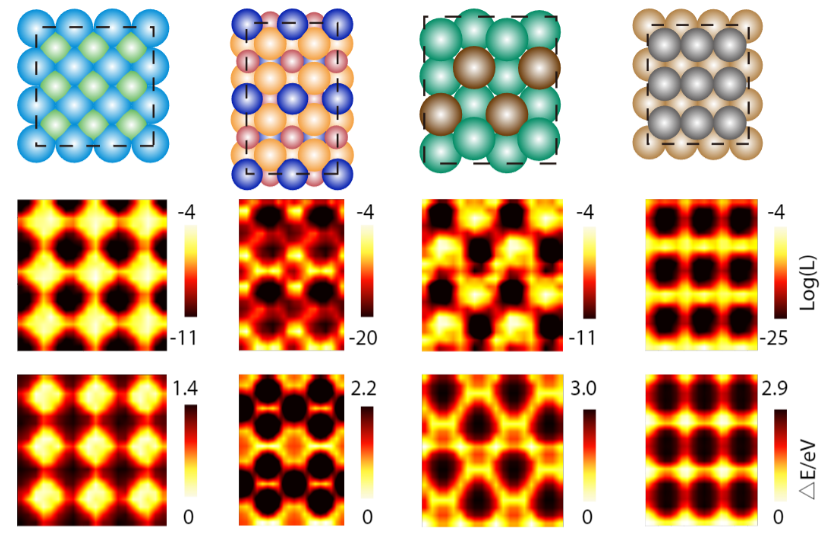
Making use of DiG, we predicted the adsorption websites for a wide range of catalyst-adsorbate methods and in contrast these predicted chances with energies obtained from DFT calculations. We discovered that DiG may discover all of the steady adsorption websites and generate adsorbate configurations which might be much like the DFT outcomes with excessive effectivity and at a low price. DiG estimates the possibilities of various adsorption configurations, in good settlement with DFT energies.
Conclusion
On this weblog, we launched DiG, a deep studying framework that goals to foretell the distribution of molecular buildings. DiG is a major development from single construction prediction towards ensemble modeling with equilibrium distributions, setting a cornerstone for connecting microscopic buildings to macroscopic properties below deep studying frameworks.
DiG includes key ML improvements that result in expressive generative fashions, which have been proven to have the capability to pattern multimodal distribution inside a given class of molecules. We have now demonstrated the flexibleness of this strategy on totally different courses of molecules (together with proteins, and so forth.), and now we have proven that particular person buildings generated on this manner are chemically lifelike. Consequently, DiG allows the event of ML methods that may pattern equilibrium distributions of molecules given acceptable coaching information.
Nonetheless, we acknowledge that significantly extra analysis is required to acquire environment friendly and dependable predictions of equilibrium distributions for arbitrary molecules. We hope that DiG conjures up extra analysis and innovation on this course, and we sit up for extra thrilling outcomes and influence from DiG and different associated strategies sooner or later.

{kind=link}