

Dec 10, 2023
(Nanowerk Highlight) The pursuit of extra environment friendly natural electronics, from OLED shows to natural photo voltaic cells, has been a permanent problem throughout supplies science. However the vastness of potential natural compounds and limitations of digital screening strategies – that are superior computational methods used to guage and predict the properties of supplies – have hindered the tempo of novel supplies discovery.
Researchers have strived to systematically navigate chemical area and precisely predict molecular properties to unlock new high-performing compounds. Nevertheless, present approaches bias the exploration towards incremental enhancements or wrestle to extract generative design ideas.
On one hand, bottom-up methods begin with identified molecular constructing blocks and mix them in several methods, enabling fast digital screening. Nevertheless, they’re biased towards incremental enhancements of present supplies moderately than wholly new households of compounds. However, top-down information mining of structural databases casts a wider web however struggles to extract actionable design pointers from the outputs.
Within the discipline of supplies science, there are two major methods for locating new supplies. On one hand, ‘bottom-up’ methods start with identified molecular constructing blocks. Consider it like utilizing LEGO bricks to create new constructions; scientists mix these blocks in numerous methods, enabling fast computerized testing of their properties. This methodology, nevertheless, tends to give attention to making small enhancements to present supplies moderately than creating utterly new ones. However, ‘top-down’ information mining entails looking via huge databases of molecular constructions to search out potential new supplies. It is akin to casting a large web to see what you’ll find, however this method usually struggles to determine sensible pointers for creating these supplies from the info it uncovers.
In the meantime, excessive computational prices constrain each approaches from exploring molecular areas as huge as nature itself. Predictive machine studying fashions have emerged as a technique to bypass costly quantum chemistry calculations. However mannequin growth is hampered by the shortage of sizable, high-quality coaching datasets with related excited state properties calculated from first ideas.
Now, researchers from EPFL have married bottom-up development with top-down information mining in a hybrid supplies discovery platform. The enabling breakthroughs are a curated dataset of 117,000 synthesized natural constructions with TDDFT-computed properties, automated identification of cross-coupling websites to generate new compounds on the fly, and quick but correct machine studying fashions to foretell key optical traits.
The findings have been printed in Superior Supplies (“Knowledge-Pushed Discovery of Natural Digital Supplies Enabled by Hybrid Prime-Down/Backside-Up Design”).
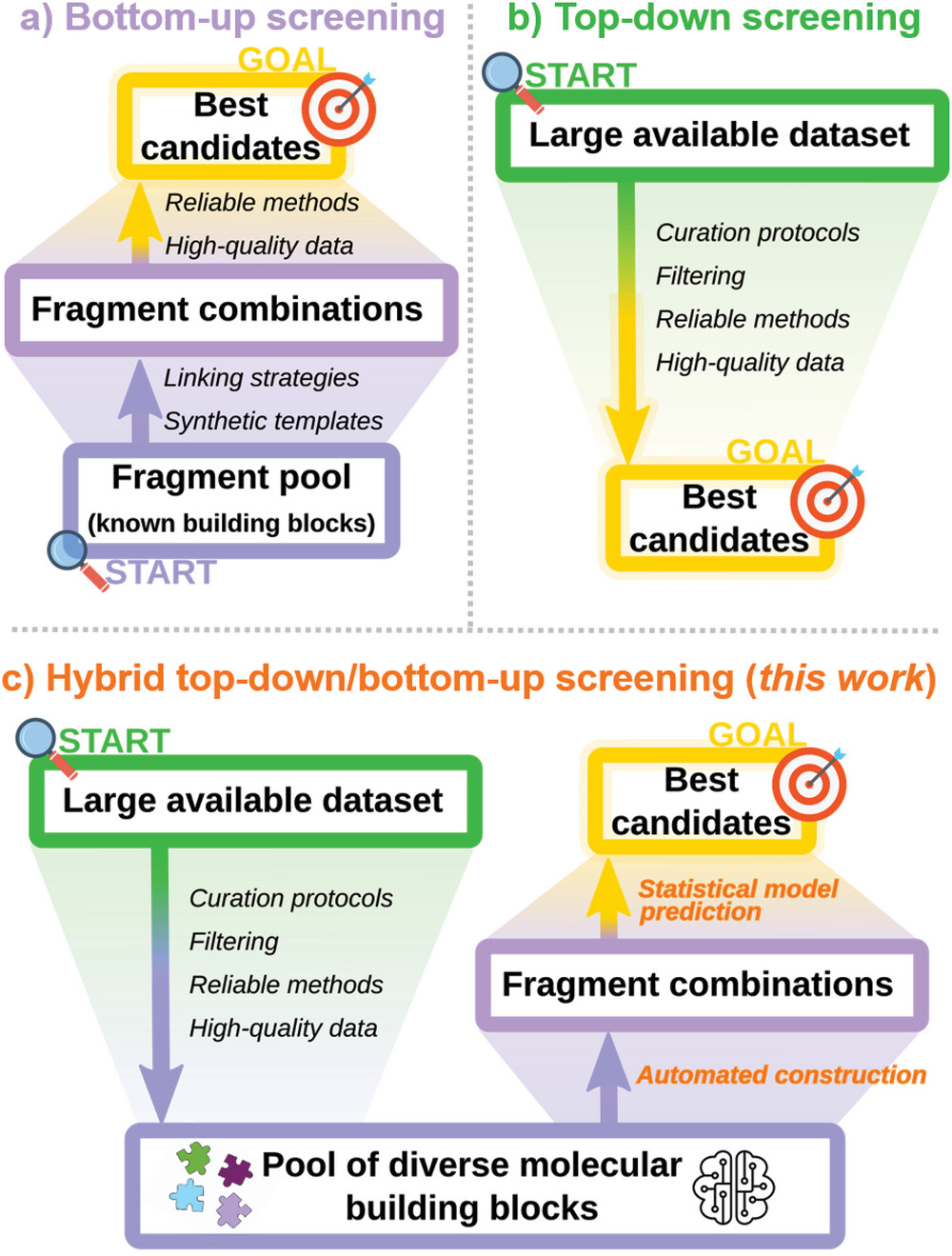 Common view of the a) bottom-up (purple) and b) top-down (inexperienced) philosophies of high-throughput digital screening, with with end-goals in yellow. Necessities for the completely different phases of a profitable screening marketing campaign are indicated in italics and datasets are proven as blocks, whose widths symbolize the datasets’ sizes relative to 1 one other. c) The hybrid screening methodology described on this work, with the union of top-down (inexperienced) and bottom-up (purple) parts enabled by statistical fashions and automatic fragment coupling (orange). (© Wiley-VCH)
Powered by this data-driven infrastructure, the researchers demonstrated ultra-large-scale screening for singlet fission candidates. From over 1,000,000 constructed donor-acceptor dimers, a whole lot of 1000’s had been flagged as having best excited state energetics for photo voltaic cells. Many characteristic uncommon molecular motifs like nitrones and furoxans which have scarcely been investigated for singlet fission until now.
Tapping the complete range of already synthesized and steady compounds debiases the search from standard constructing blocks. And by coaching fashions on quantum-accurate information spanning identified chemical area, dependable predictions unlock digital screening at unprecedented scales.
The researchers extracted over 167,000 crystal constructions from the Cambridge Structural Database, representing the most important public compilation of characterised artificial compounds. In depth curation eradicated duplicates, errors, and nonsensically bonded moieties.
The ultimate filtered set of 117K compounds offers a structurally various palette spanning identified natural chemistry far past standard molecular datasets. In contrast even to the 134K small molecules within the established QM9 set, this covers extra parts throughout a higher vary of sizes with out constraints.
From every refined crystal construction, a set of digital properties was calculated utilizing density practical approximations. This contains frontier orbital energies, a number of low-lying singlet and triplet excitation ranges, and quantitative descriptors of transition character. Collectively these properties inform exciton conduct and cost switch character, underlying functions from OLED emitters to natural photovoltaics.
To remodel static molecule snapshots into versatile constructing blocks, the researchers developed a topology-based device that flags potential carbon websites appropriate for cross-coupling reactions. Specializing in unsaturated C-H positions, they enumerate the native chemical environments to determine symmetry-equivalent teams.
This protocol mimics retrosynthetic evaluation to decode websites the place fragments might be stitched collectively by established carbon-carbon bond formations. Codifying the power to couple straight extracts synthesis-relevant data from 3D structural information. It additionally permits algorithmic meeting of derivatives not initially current within the dataset.
To bypass the necessity for costly excited state calculations on each new hypothetical compound, the crew skilled machine studying fashions utilizing the precomputed information. The inputs illustration integrated geometry-dependent London and Axilrod-Teller-Muto potentials—physics-inspired atomic pairwise and triplet phrases.
The fashions achieved exceptional accuracy in predicting singlet and triplet excitation energies for various natural constructions. This required including a number of thousand computer-generated dimers to the coaching information for enough illustration of bonded molecular pairs. The fashions recapitulate quantum chemistry with a fraction of the computational expense, forecasting optical properties in a flash.
The researchers demonstrated the capabilities of their hybrid discovery infrastructure by developing and screening over a million donor-acceptor dimers for intramolecular singlet fission candidacy. Recognized singlet fission supplies are scarce, delicate, undergo from low triplet vitality, and derive principally from oligoacene derivatives.
By stipulating complementary design standards primarily based on frontier orbital patterns and excitation energetics, the crew rationally chosen donor and acceptor fragments with acceptable traits from inside their molecular database. Automated coupling of those 1000’s of constructing blocks adopted by ML-accelerated screening yielded nearly 560,000 structurally distinct dimers with best thermodynamic signatures for singlet fission and triplet energies suited to frequent photovoltaic absorbers like silicon and CdTe.
Many comprise not often investigated acceptor teams like nitrones and furoxans that maintain untapped promise for singlet fission, as corroborated via substructure evaluation. Beginning straight from molecules already efficiently synthesized avoids reliance on standard intuitions about productive chemical subunits. This highlights how data-driven compound technology powered by predictive modeling offers an agnostic avenue to uncharted molecular territories with desired properties.
Common view of the a) bottom-up (purple) and b) top-down (inexperienced) philosophies of high-throughput digital screening, with with end-goals in yellow. Necessities for the completely different phases of a profitable screening marketing campaign are indicated in italics and datasets are proven as blocks, whose widths symbolize the datasets’ sizes relative to 1 one other. c) The hybrid screening methodology described on this work, with the union of top-down (inexperienced) and bottom-up (purple) parts enabled by statistical fashions and automatic fragment coupling (orange). (© Wiley-VCH)
Powered by this data-driven infrastructure, the researchers demonstrated ultra-large-scale screening for singlet fission candidates. From over 1,000,000 constructed donor-acceptor dimers, a whole lot of 1000’s had been flagged as having best excited state energetics for photo voltaic cells. Many characteristic uncommon molecular motifs like nitrones and furoxans which have scarcely been investigated for singlet fission until now.
Tapping the complete range of already synthesized and steady compounds debiases the search from standard constructing blocks. And by coaching fashions on quantum-accurate information spanning identified chemical area, dependable predictions unlock digital screening at unprecedented scales.
The researchers extracted over 167,000 crystal constructions from the Cambridge Structural Database, representing the most important public compilation of characterised artificial compounds. In depth curation eradicated duplicates, errors, and nonsensically bonded moieties.
The ultimate filtered set of 117K compounds offers a structurally various palette spanning identified natural chemistry far past standard molecular datasets. In contrast even to the 134K small molecules within the established QM9 set, this covers extra parts throughout a higher vary of sizes with out constraints.
From every refined crystal construction, a set of digital properties was calculated utilizing density practical approximations. This contains frontier orbital energies, a number of low-lying singlet and triplet excitation ranges, and quantitative descriptors of transition character. Collectively these properties inform exciton conduct and cost switch character, underlying functions from OLED emitters to natural photovoltaics.
To remodel static molecule snapshots into versatile constructing blocks, the researchers developed a topology-based device that flags potential carbon websites appropriate for cross-coupling reactions. Specializing in unsaturated C-H positions, they enumerate the native chemical environments to determine symmetry-equivalent teams.
This protocol mimics retrosynthetic evaluation to decode websites the place fragments might be stitched collectively by established carbon-carbon bond formations. Codifying the power to couple straight extracts synthesis-relevant data from 3D structural information. It additionally permits algorithmic meeting of derivatives not initially current within the dataset.
To bypass the necessity for costly excited state calculations on each new hypothetical compound, the crew skilled machine studying fashions utilizing the precomputed information. The inputs illustration integrated geometry-dependent London and Axilrod-Teller-Muto potentials—physics-inspired atomic pairwise and triplet phrases.
The fashions achieved exceptional accuracy in predicting singlet and triplet excitation energies for various natural constructions. This required including a number of thousand computer-generated dimers to the coaching information for enough illustration of bonded molecular pairs. The fashions recapitulate quantum chemistry with a fraction of the computational expense, forecasting optical properties in a flash.
The researchers demonstrated the capabilities of their hybrid discovery infrastructure by developing and screening over a million donor-acceptor dimers for intramolecular singlet fission candidacy. Recognized singlet fission supplies are scarce, delicate, undergo from low triplet vitality, and derive principally from oligoacene derivatives.
By stipulating complementary design standards primarily based on frontier orbital patterns and excitation energetics, the crew rationally chosen donor and acceptor fragments with acceptable traits from inside their molecular database. Automated coupling of those 1000’s of constructing blocks adopted by ML-accelerated screening yielded nearly 560,000 structurally distinct dimers with best thermodynamic signatures for singlet fission and triplet energies suited to frequent photovoltaic absorbers like silicon and CdTe.
Many comprise not often investigated acceptor teams like nitrones and furoxans that maintain untapped promise for singlet fission, as corroborated via substructure evaluation. Beginning straight from molecules already efficiently synthesized avoids reliance on standard intuitions about productive chemical subunits. This highlights how data-driven compound technology powered by predictive modeling offers an agnostic avenue to uncharted molecular territories with desired properties.
Common view of the a) bottom-up (purple) and b) top-down (inexperienced) philosophies of high-throughput digital screening, with with end-goals in yellow. Necessities for the completely different phases of a profitable screening marketing campaign are indicated in italics and datasets are proven as blocks, whose widths symbolize the datasets’ sizes relative to 1 one other. c) The hybrid screening methodology described on this work, with the union of top-down (inexperienced) and bottom-up (purple) parts enabled by statistical fashions and automatic fragment coupling (orange). (© Wiley-VCH)
Powered by this data-driven infrastructure, the researchers demonstrated ultra-large-scale screening for singlet fission candidates. From over 1,000,000 constructed donor-acceptor dimers, a whole lot of 1000’s had been flagged as having best excited state energetics for photo voltaic cells. Many characteristic uncommon molecular motifs like nitrones and furoxans which have scarcely been investigated for singlet fission until now.
Tapping the complete range of already synthesized and steady compounds debiases the search from standard constructing blocks. And by coaching fashions on quantum-accurate information spanning identified chemical area, dependable predictions unlock digital screening at unprecedented scales.
The researchers extracted over 167,000 crystal constructions from the Cambridge Structural Database, representing the most important public compilation of characterised artificial compounds. In depth curation eradicated duplicates, errors, and nonsensically bonded moieties.
The ultimate filtered set of 117K compounds offers a structurally various palette spanning identified natural chemistry far past standard molecular datasets. In contrast even to the 134K small molecules within the established QM9 set, this covers extra parts throughout a higher vary of sizes with out constraints.
From every refined crystal construction, a set of digital properties was calculated utilizing density practical approximations. This contains frontier orbital energies, a number of low-lying singlet and triplet excitation ranges, and quantitative descriptors of transition character. Collectively these properties inform exciton conduct and cost switch character, underlying functions from OLED emitters to natural photovoltaics.
To remodel static molecule snapshots into versatile constructing blocks, the researchers developed a topology-based device that flags potential carbon websites appropriate for cross-coupling reactions. Specializing in unsaturated C-H positions, they enumerate the native chemical environments to determine symmetry-equivalent teams.
This protocol mimics retrosynthetic evaluation to decode websites the place fragments might be stitched collectively by established carbon-carbon bond formations. Codifying the power to couple straight extracts synthesis-relevant data from 3D structural information. It additionally permits algorithmic meeting of derivatives not initially current within the dataset.
To bypass the necessity for costly excited state calculations on each new hypothetical compound, the crew skilled machine studying fashions utilizing the precomputed information. The inputs illustration integrated geometry-dependent London and Axilrod-Teller-Muto potentials—physics-inspired atomic pairwise and triplet phrases.
The fashions achieved exceptional accuracy in predicting singlet and triplet excitation energies for various natural constructions. This required including a number of thousand computer-generated dimers to the coaching information for enough illustration of bonded molecular pairs. The fashions recapitulate quantum chemistry with a fraction of the computational expense, forecasting optical properties in a flash.
The researchers demonstrated the capabilities of their hybrid discovery infrastructure by developing and screening over a million donor-acceptor dimers for intramolecular singlet fission candidacy. Recognized singlet fission supplies are scarce, delicate, undergo from low triplet vitality, and derive principally from oligoacene derivatives.
By stipulating complementary design standards primarily based on frontier orbital patterns and excitation energetics, the crew rationally chosen donor and acceptor fragments with acceptable traits from inside their molecular database. Automated coupling of those 1000’s of constructing blocks adopted by ML-accelerated screening yielded nearly 560,000 structurally distinct dimers with best thermodynamic signatures for singlet fission and triplet energies suited to frequent photovoltaic absorbers like silicon and CdTe.
Many comprise not often investigated acceptor teams like nitrones and furoxans that maintain untapped promise for singlet fission, as corroborated via substructure evaluation. Beginning straight from molecules already efficiently synthesized avoids reliance on standard intuitions about productive chemical subunits. This highlights how data-driven compound technology powered by predictive modeling offers an agnostic avenue to uncharted molecular territories with desired properties.

By
Michael
Berger
– Michael is creator of three books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Expertise,
Nanotechnology: The Future is Tiny, and
Nanoengineering: The Expertise and Instruments Making Expertise Invisible
Copyright ©
Nanowerk LLC
Turn into a Highlight visitor creator! Be a part of our giant and rising group of visitor contributors. Have you ever simply printed a scientific paper or produce other thrilling developments to share with the nanotechnology group? Right here is how you can publish on nanowerk.com.

{kind=link}